
Japanese–European Research Team Discovers Novel Genetic Mitochondrial Disorder
Team of Japanese and European scientists identify a novel genetic mitochondrial disorder by analyzing DNA samples from three distinct families
The list of known genetic mitochondrial disorders is ever-growing, and ongoing research continues to identify new disorders in this category. In an article recently published in Brain, a Japanese-European team of scientists, including researchers from Fujita Health University, describe mutations in the LIG3 gene, which plays a crucial role in mitochondrial DNA replication. These mutations cause a previously unknown syndrome characterized by gut dysmotility, leukoencephalopathy, and neuromuscular abnormalities.
DNA ligase proteins, which facilitate the formation of bonds between separate strands of DNA, play critical roles in the replication and maintenance of DNA. The human genome encodes three different DNA ligase proteins, but only one of those proteins—DNA ligase III (LIG3)—is expressed in mitochondria. LIG3 is therefore crucial for mitochondrial health, and inactivation of the homologous protein in mice causes profound mitochondrial dysfunction and early embryonic mortality. In an article recently published in the peer-reviewed journal Brain, a team of European and Japanese scientists, led by Dr. Mariko Taniguchi-Ikeda from Fujita Health University Hospital, describes a set of seven patients with a novel mitochondrial disorder caused by biallelic variants in the gene that encodes the LIG3 protein, called the “LIG3” gene. Their report provides a description of the patients’ symptoms and a mechanistic exploration of the mutations’ effects.
For Dr. Taniguchi-Ikeda, the investigation began with her desire to help a young patient. “I wanted to make a distinct clinical and genetic diagnosis for the affected patient,” she explains, “because his elder brother had passed away and the surviving boy was referred to my outpatient ward for detailed genetic tests.” By performing whole-exome sequencing of DNA from the surviving patient, Dr. Taniguchi-Ikeda discovered that he had inherited a p.P609L LIG3 variant from his father and a p.R811Ter LIG3 variant from his mother. The parents had kept the deceased brother’s dried umbilical cord, and by analyzing DNA extracted from that source, Dr. Taniguchi-Ikeda confirmed that the brother had carried the same LIG3 variants.
Having detected a novel genetic mitochondrial disorder, Dr. Taniguchi-Ikeda wished to conduct further research by identifying other patients with pathogenic LIG3 variants. She could find no other such cases in Japan, but through a collaboration with Dr. Makiko Tsutsumi from Fujita Health University and researchers in Europe, including Professor Elena Bonora from the University of Bologna and Professor Roberto De Giorgio from the University of Ferrara, she learned of two European families also affected by such variants. One was an Italian family in which three brothers had all inherited a p.K537N variant from their father and a p.G964R variant from their mother, and the other was a Dutch family in which two daughters had inherited a p.R267Ter variant from their father and a p.C999Y variant from their mother.
These patients experienced a complex syndrome involving severe gut dysmotility and neurologic abnormalities as the most consistently observed clinical signs. The neurologic abnormalities included leukoencephalopathy, epilepsy, migraine, stroke-like episodes, and neurogenic bladder. The prominent changes in the gut were decreased myenteric neuron counts and elevated fibrosis and elastin levels. Muscle pathology assessments revealed decreased staining intensities for cytochrome C oxidase.
To better characterize how the patients’ LIG3 mutations could lead to such phenotypes, the researchers conducted experiments both in vitro and on zebrafish. The in vitro experiments with patient-derived fibroblasts showed that the mutations resulted in reduced LIG3 protein levels and diminished ligase activity. The consequent deficits in mitochondrial DNA maintenance would do much to explain the patients’ presentations. Experiments with zebrafish showed that disrupting the lig3 gene produced brain alterations and gut transit impairments analogous to those observed in the patients.
The study brings to light a novel disorder resulting from disruption of a gene that plays a critical role in the maintenance of mitochondrial DNA. In describing the importance of these findings, Dr. Taniguchi-Ikeda concludes, “Our study may facilitate efforts to diagnose patients with mitochondrial diseases. Our findings will also be beneficial to future investigations into the mitochondrial DNA repair system.”
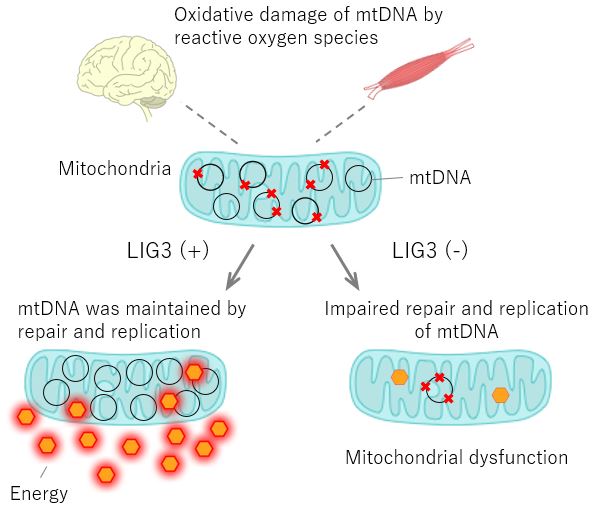
A Japanese-European team of scientists described novel mutations in the LIG3 gene, which plays a key role in mitochondrial DNA replication. Normally, mitochondrial DNA is repaired and replicated by LIG3 activity, but if the gene contains mutations, enzymes necessary for energy production are not produced, potentially leading to central nervous system symptoms and muscle defects.
The list of known genetic mitochondrial disorders is ever-growing, and ongoing research continues to identify new disorders in this category. In an article recently published in Brain, a Japanese-European team of scientists, including researchers from Fujita Health University, describe mutations in the LIG3 gene, which plays a crucial role in mitochondrial DNA replication. These mutations cause a previously unknown syndrome characterized by gut dysmotility, leukoencephalopathy, and neuromuscular abnormalities.
DNA ligase proteins, which facilitate the formation of bonds between separate strands of DNA, play critical roles in the replication and maintenance of DNA. The human genome encodes three different DNA ligase proteins, but only one of those proteins—DNA ligase III (LIG3)—is expressed in mitochondria. LIG3 is therefore crucial for mitochondrial health, and inactivation of the homologous protein in mice causes profound mitochondrial dysfunction and early embryonic mortality. In an article recently published in the peer-reviewed journal Brain, a team of European and Japanese scientists, led by Dr. Mariko Taniguchi-Ikeda from Fujita Health University Hospital, describes a set of seven patients with a novel mitochondrial disorder caused by biallelic variants in the gene that encodes the LIG3 protein, called the “LIG3” gene. Their report provides a description of the patients’ symptoms and a mechanistic exploration of the mutations’ effects.
For Dr. Taniguchi-Ikeda, the investigation began with her desire to help a young patient. “I wanted to make a distinct clinical and genetic diagnosis for the affected patient,” she explains, “because his elder brother had passed away and the surviving boy was referred to my outpatient ward for detailed genetic tests.” By performing whole-exome sequencing of DNA from the surviving patient, Dr. Taniguchi-Ikeda discovered that he had inherited a p.P609L LIG3 variant from his father and a p.R811Ter LIG3 variant from his mother. The parents had kept the deceased brother’s dried umbilical cord, and by analyzing DNA extracted from that source, Dr. Taniguchi-Ikeda confirmed that the brother had carried the same LIG3 variants.
Having detected a novel genetic mitochondrial disorder, Dr. Taniguchi-Ikeda wished to conduct further research by identifying other patients with pathogenic LIG3 variants. She could find no other such cases in Japan, but through a collaboration with Dr. Makiko Tsutsumi from Fujita Health University and researchers in Europe, including Professor Elena Bonora from the University of Bologna and Professor Roberto De Giorgio from the University of Ferrara, she learned of two European families also affected by such variants. One was an Italian family in which three brothers had all inherited a p.K537N variant from their father and a p.G964R variant from their mother, and the other was a Dutch family in which two daughters had inherited a p.R267Ter variant from their father and a p.C999Y variant from their mother.
These patients experienced a complex syndrome involving severe gut dysmotility and neurologic abnormalities as the most consistently observed clinical signs. The neurologic abnormalities included leukoencephalopathy, epilepsy, migraine, stroke-like episodes, and neurogenic bladder. The prominent changes in the gut were decreased myenteric neuron counts and elevated fibrosis and elastin levels. Muscle pathology assessments revealed decreased staining intensities for cytochrome C oxidase.
To better characterize how the patients’ LIG3 mutations could lead to such phenotypes, the researchers conducted experiments both in vitro and on zebrafish. The in vitro experiments with patient-derived fibroblasts showed that the mutations resulted in reduced LIG3 protein levels and diminished ligase activity. The consequent deficits in mitochondrial DNA maintenance would do much to explain the patients’ presentations. Experiments with zebrafish showed that disrupting the lig3 gene produced brain alterations and gut transit impairments analogous to those observed in the patients.
The study brings to light a novel disorder resulting from disruption of a gene that plays a critical role in the maintenance of mitochondrial DNA. In describing the importance of these findings, Dr. Taniguchi-Ikeda concludes, “Our study may facilitate efforts to diagnose patients with mitochondrial diseases. Our findings will also be beneficial to future investigations into the mitochondrial DNA repair system.”
A Japanese-European team of scientists described novel mutations in the LIG3 gene, which plays a key role in mitochondrial DNA replication. Normally, mitochondrial DNA is repaired and replicated by LIG3 activity, but if the gene contains mutations, enzymes necessary for energy production are not produced, potentially leading to central nervous system symptoms and muscle defects.
Reference
Title of original paper
Biallelic variants in LIG3 cause a novel mitochondrial neurogastrointestinal encephalomyopathyJournal
BrainDOI
10.1093/brain/awab056About Dr. Mariko Taniguchi-Ikeda
Dr. Mariko Taniguchi-Ikeda, MD, PhD, is a pediatric physician and researcher who is affiliated with the Department of Clinical Genetics at Fujita Health University Hospital as an Associate Professor. She is also affiliated with the Institute for Comprehensive Medical Science at Fujita Health University and the Department of Pediatrics at the Kobe University Graduate School of Medicine. Her research interests include molecular genetics, congenital muscular dystrophy, and pediatrics.
Funding information
This study was funded by a Telethon Grant, the city of Kobe, the Dutch Cancer Foundation, the Italian Ministry of Health, and the University of Ferrara.
mtani@fujita-hu.ac.jp| taniguchi_mariko@me.com
Media contact
Mariko Taniguchi-Ikedamtani@fujita-hu.ac.jp| taniguchi_mariko@me.com